[VIP第1年] 指数:3
[VIP第1年] 指数:3
荧光PCR技术是目前临床基因检测的优先方案,因其精确、快速、简单且成本较低,荧光PCR仪一次反应可以容纳96个或者384个样品,检测通量要高于FISH、IHC等技术;在除了高通量测序以外的技术中,荧光PCR也是检测DNA变异类型**多的方法。此外,荧光PCR法技术壁垒较低,易于生产,广东伴随诊断口碑推荐。荧光PCR的弱点也很明显,由于原理所限,荧光PCR法只能检测常见位点的突变,无法覆盖该基因的所有碱基,这可能导致忽略某些稀有的突变得到假阴性的结果,从而造成误判。FISH技术目前只用于检测基因扩增和融合基因,结果可直接在显微镜下观察到,过程直观操作简单,是产品数量*次于荧光PCR的方法。这种方法的局限性在于只适用于检测单个基因片段的融合或扩增。并且需要较为专业的判读技术。二代测序技术的出现为**的易感基因检测、伴随诊断,广东伴随诊断口碑推荐、个性化用药等提供了更佳的技术支撑和选择,尤其是基于NGS的aizhengpanel检测使得该领域检测可以更加快速和低廉,广东伴随诊断口碑推荐,达到了同时检测若干个基因和突变位点的目的。迈杰转化医学拥有专业的病理医生提供相应的阅片或远程病理阅片服务。广东伴随诊断口碑推荐
基于此研究的数据支持,日本厚生劳动省批准了艾德生物ROS1试剂盒作为克唑替尼的伴随诊断试剂,这是国内企业较早在海外获批的伴随诊断试剂。同时,艾德生物ROS1试剂盒已正式获批进入日本全国医保,可使日本每年新增的8万多名NSCLC患者获得ROS1伴随诊断检测和药物疗治的机会。克唑替尼亚太临床研究日本项目负责人,日本国家ai症中心肺ai项目LC-SCRUM负责人,,艾德生物ROS1试剂盒具有高灵敏度,高准确度,高便捷性,适用样本类型广等特点,并且可以提供更多融合分型信息,为临床提供疗治依据。艾德生物ROS1试剂盒在日本获批上市并进入医保,标志着中国创造的伴随诊断产品已在发达国家医疗市场崭露头角,中国企业已成为辉瑞等全球知I名药企跨国合作的新宠儿。艾德生物专注于**精细医疗的伴随检测领域,开发出我国首批获得CFDA和欧盟CE认证的**精细医疗伴随诊断产品,在全球知I名的欧洲分子基因诊断质量联盟(EMQN)室间质评中,艾德生物产品使用率连续三年保持***。艾德生物技术和产品已获得国内外***认可,成为Pfizer、Merck、Illumina、AstraZeneca、Boehringer-Ingelheim等多家跨国企业的战略合作伙伴。目前全球50多个国家和地区的数百家大中型医院选择了艾德产品。湖南推荐伴随诊断口碑推荐迈杰多平台的研究优势以及多组学数据的挖掘能力,促进产学研医结合,加速项目成果转化,创新科技产品研发。
是较早基于NGS技术上市的伴随诊断产品,该产品可用于检测患者BRCA基因是否缺陷;2017年11月,FDA又批准了FoundationMedicine公司的产品FoundationOneCDx(F1CDx),该产品可以检测300多个**相关基因,可用于所有类型的实体瘤,在**精细医疗领域中实现了重大突破。FDA也先后发布了一些指导性的意见来指导这个行业的发展:国内的伴随诊断则起步较晚。2015年3月,科技部召开了国家***“精细医学战略**会议”,会议计划,在2030年前要在精细医疗领域内投入600亿元;2015年7月,卫计委个体化医学检测技术**委员会制订了《**个体化疗治检测技术指南(试行)》和《药物代谢酶和药物作用靶点基因检测技术指南(试行)》,要求实现**精细医疗用药基因检测标准化和规范化。
ExtendedRASPanel可帮助鉴别适用于安进公司Vectibix的结直肠*患者圣迭戈——2017年6月29日——Illumina公司(纳斯达克股票代码:ILMN)JIN天宣布ExtendedRASPanel已获美国食品药品监督管理局(FDA)批准发布,这一新一代测序(NGS)套件符合由美国临床病理学学会(ASCP)、美国病理学家协会(CAP)、美国分子病理学会(AMP)以及美国临床**学会(ASCO)***发布的关于结直肠*评估的指南。此套件用于IlluminaMiSeqDx?系统,可以让美国的实验室有能力帮助临床医师鉴别哪些患者适用于使用Vectibix?(帕尼单抗)***转移性结直肠*。作为较早由FDA批准与FOLFOX共同用于*****野生型RAS转移性结直肠*(mCRC)患者的单克隆抗表皮生长因子受体(EGFR)抗体,对于这些患者来说,Vectibix**了一种全新的***选择。在Vectibix与FOLFOX共同作用于野生型RASmCRC患者的过程中,观察到其总生存率和无进展生存期同时提高,这更加凸显出利用生物标记进行筛查来优化*症***计划制定的重要性。“我们与安进公司共同开发了一个伴随诊断检测套件,该套件审视了KRAS和NRAS基因中的56个变异,能够在单次检测中确定突变情况。通过ExtendedRASPanel。迈杰转化医学同时拥有符合GMP和ISO 13485的要求的GMP生产车间及仓储,可以充分满足客户的需求。
2.策略二:当没有批准的IVD用于该生物标志物的检测时,应该使用实验室自建检测(Laboratory-DevelopedTests,LDTs)进行患者筛选,如果药物开发后期有IVD获批,应该进行桥接研究(BridgingStudy)验证LDT与IVD具有非常类似的检测性能(VerySimilarPerformanceCharacteristics)。例如曲妥珠单抗在乳腺*临床试验时使用临床试验检测(ClinicalTrialAssay,CTA)进行患者筛选,后来通过桥接研究证明了HercepTest与CTA检测结果的一致性。3.策略三:如果药物开发初期,用于患者筛选的生物标志物不明确,临床试验不需要进行基于生物标志物分组的队列研究。一旦生物标志物明确,应该进行回顾性分析(RetrospectiveAnalysis)来评价生物标志物。如果生物标志物阳性队列的有效性或安全性(Efficacy/Safety)优于阴性队列,那么药物说明书(DrugLabel)中应该增加生物标志物的相关信息。例如KRASmutationkits和其伴随药物西妥昔单抗(Cetuximab),就是通过这种策略开发的。 迈杰转化医学为创新药企开展全球多中心临床试验研究,提供中心实验室检测及伴随诊断开发服务。湖南推荐伴随诊断口碑推荐
迈杰拥有StarLIMS实验室信息化管理系统,中心实验室获得了中国CNAS ISO 17025实验室认可、美国CAP认证。广东伴随诊断口碑推荐
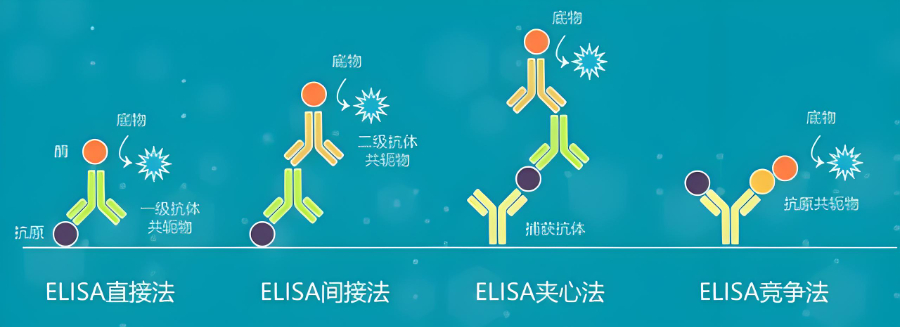
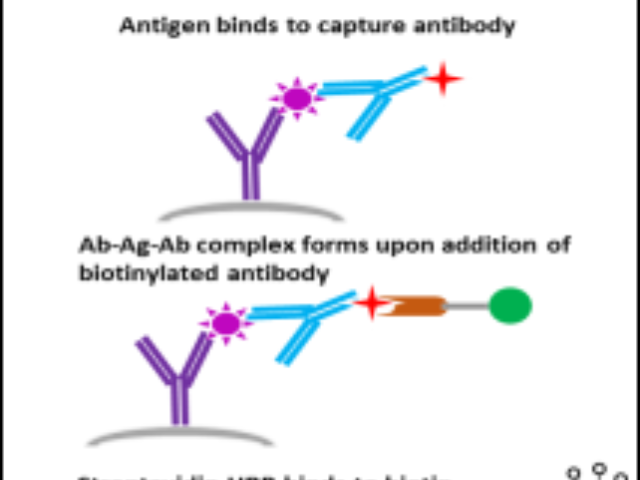
公司已看到伴随诊断的发展潜力,开始布局相关业务,和信达生物共同开拓伴随诊断市场,与阿斯利康 就HRD伴随诊断试剂盒(CDX)合作,与阿斯利康签署战略合作协议,共同开发同源重组缺陷(HRD)**伴随诊断项目,并加入HRD检测公益联盟。1998年,FDA批准了HercepTest用于识别能从曲妥珠单抗(Trastuzumab)***中获益的人群,这是***个FDA批准的伴随诊断,截止2021年6月FDA已经批准了45个伴随诊断,主要基于免疫组织化学(Immunohistochemistry,IHC)、荧光原位杂交(Fluorescence In Situ Hybridization,FISH)、显色原位杂交(Chromogenic In Situ Hybridization,CISH)、聚合酶链式反应(Polymerase Chain Reaction,PCR)、下一代测序(Next Generation Sequencing,NGS)等方法开发,表2列举了基于这些方法开发的伴随诊断[4,5]:广东伴随诊断口碑推荐
文章来源地址: http://yyby.chanpin818.com/swzp/swzdsj/deta_13920371.html
免责声明: 本页面所展现的信息及其他相关推荐信息,均来源于其对应的用户,本网对此不承担任何保证责任。如涉及作品内容、 版权和其他问题,请及时与本网联系,我们将核实后进行删除,本网站对此声明具有最终解释权。