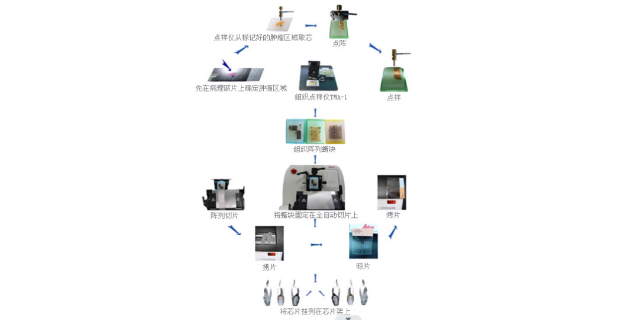
[VIP第1年] 指数:3
[VIP第1年] 指数:3
除了比较基因组学研究,泛基因组分析也是近年来备受关注的研究方向。泛基因组包括了一个物种内所有基因组水平发生的变异。借助生物信息学技术手段,我们可以在基因组数据中挖掘大量的潜在基因,包括了显性基因和隐性基因,这为我们解释细菌的多样性和适应性提供了新的视角。此外,泛基因组的研究还有助于理解细菌内多样性的形成和演化特点,深入探究细菌在微生物群体中的生态意义和功能。综上所述,基于生物信息学技术手段下获得的细菌基因组完成图序列开展基因功能注释、比较基因组学以及泛基因组的研究,为我们揭示了细菌的多样性、进化规律和适应策略,为微生物学研究提供了重要的理论基础和实践指导。随着技术的不断进步和研究方法的不断丰富,相信细菌基因组学的研究将继续取得新的突破和进展,为微生物资源开发和生物技术应用提供更多的支持和帮助。 用于监测和治理环境污染,如生物修复和生物监测等。核酸的分离提取

在细菌基因组研究中,从头测序是一项至关重要的工作,它为我们打开了深入了解细菌世界的大门。通过对序列进行拼接和组装,我们能够逐步构建出完整的细菌基因组序列,这一过程充满了挑战与惊喜。当我们着手进行从头测序时,首先面临的是海量的原始序列数据。这些数据就像是无数的拼图碎片,等待着我们去正确地组合和拼接。为了实现这一目标,科学家们运用了一系列复杂而精巧的技术和算法。初始阶段,测序仪器会产生大量短的DNA序列片段,这些片段可能只有几百个碱基对长。接下来的关键步骤就是将这些片段进行比对和拼接。这需要强大的计算能力和精确的算法支持,以确保每一个片段都能被准确地放置在基因组的正确位置上。基因测序成本通过比较不同细菌物种的基因组序列,分析它们之间的差异和相似性,揭示细菌的进化关系和功能特征。
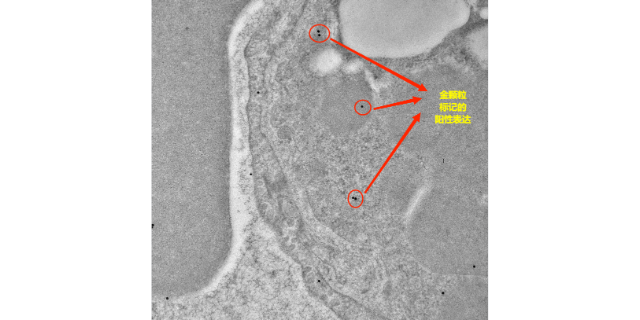
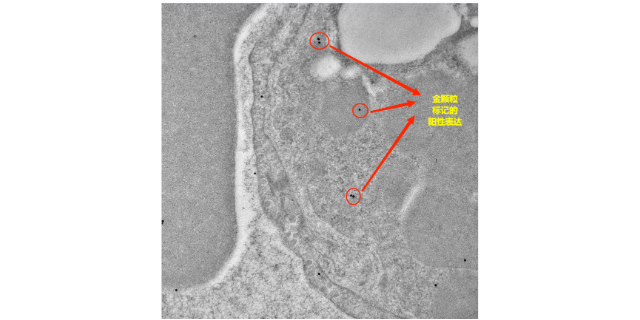
细菌基因组,虽然相对简单,但却蕴含着决定细菌特性和行为的关键信息。当细菌群体中的基因组发生变异时,就像是一场悄然进行的变革。群体变异的发生有着多种原因。首先,细菌具有极高的繁殖速度,在短时间内可以产生大量的后代。在这个过程中,DNA复制可能会出现一些错误,而这些错误如果得以传递和积累,就会导致基因组的变异。其次,环境因素的压力也是促使细菌基因组发生群体变异的重要动力。例如,当细菌面临的选择压力时,一些能够产生抗药性变异的细菌就会脱颖而出,在群体中逐渐占据优势。
除了基因突变,拷贝数变异也是常见的基因组变异形式之一。拷贝数变异是指某一段基因序列的拷贝数目发生变化,造成基因组中特定基因的拷贝数增加或减少。这种变异可能导致基因的表达水平发生变化,进而影响生物体的表型特征。染色体结构变异是指染色体的结构发生改变,例如染色体片段的缺失、重排、倒位等。这种变异不仅可以导致基因的表达发生改变,还可能影响染色体的稳定性和遗传信息的传递。基因组变异在生物的进化中起着非常重要的作用。通过基因组变异,生物体可以产生新的基因型和表型,增加生物种群的遗传变异性,从而适应不同的环境压力。在进化过程中,基因组变异是生物适应环境的关键驱动力之一。细菌基因组大小可以在几百万到数百万个碱基对之间变化。

一旦成功获得了该细菌菌种的基因组序列,其意义是巨大的。我们可以深入探究细菌的基因组成、功能以及进化历程。了解细菌所具有的各种基因,包括与代谢、致病性、耐药性等相关的基因,为疾病诊断和提供重要依据。通过对不同细菌菌种的基因组序列进行比较,我们还能发现物种之间的差异和相似性,进一步揭示细菌进化的规律和机制。这对于理解细菌的适应性和多样性具有关键意义。从头测序的过程也是一个不断探索和发现的过程。在这个过程中,我们可能会遇到前所未有的基因结构和功能,为生物学领域带来新的启发和研究方向。细菌基因组包括染色体和质粒上的 DNA。细菌固体培养基
细菌在环境中起着重要的作用,通过研究细菌基因组可以了解它们在环境中的分布和功能。核酸的分离提取
在生物信息学中,有许多工具可以用于预测蛋白质的结构域。以下是一些常用的工具:InterProScan:InterProScan是一个整合了多个结构域预测数据库的工具,包括InterPro、Pfam、PRINTS、PROSITE等,可以对蛋白序列进行的结构域预测。SMART (Simple Modular Architecture Research Tool):SMART是一个基于结构域信息的工具,可以预测蛋白质中存在的功能域、结构域和域间距。用户可以输入蛋白序列进行SMART搜索,获取预测的结构域信息。Pfam:Pfam是一个使用的蛋白质家族数据库,其中包含了许多已知的蛋白质结构域信息。通过Pfam数据库,可以对蛋白序列进行结构域预测和家族分类。PROSITE:PROSITE是一个包含了各种蛋白质结构域模式和保守序列模式的数据库,可以利用PROSITE进行蛋白质结构域的检测和预测。CDD (Conserved Domain Database):CDD是NCBI提供的一个用于蛋白结构域分析的数据库,包含了结构域和功能域的信息。可以在NCBI的网站上进行CDD搜索和分析。HMMER:HMMER是一种基于隐藏马尔可夫模型(HMM)的工具,可以用于蛋白结构域的预测和序列比对。通过HMMER可以对蛋白序列中可能存在的结构域进行识别和分析。核酸的分离提取
文章来源地址: http://yyby.chanpin818.com/swzp/qtswzp/deta_23663689.html
免责声明: 本页面所展现的信息及其他相关推荐信息,均来源于其对应的用户,本网对此不承担任何保证责任。如涉及作品内容、 版权和其他问题,请及时与本网联系,我们将核实后进行删除,本网站对此声明具有最终解释权。